|
|

|
 |
|
 |
|
|
|
|
|
 |
|
 |
|
|
|
|
|
Research Highlights
protein aggregation
|
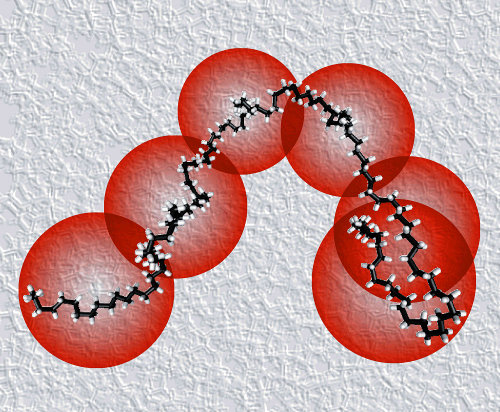
We developed a highly coarse grained model
to study the aggrgation of α-synuclein, the protein associated with
Parkinson's disease
[1],
into ordered structures.
We represent proteins
as soft bodies of various shapes
with attractive patches on their surfaces.
To simulate dynamics we have implemented a Brownian Dynamics algorithm
for the translational and rotational motion
[2].
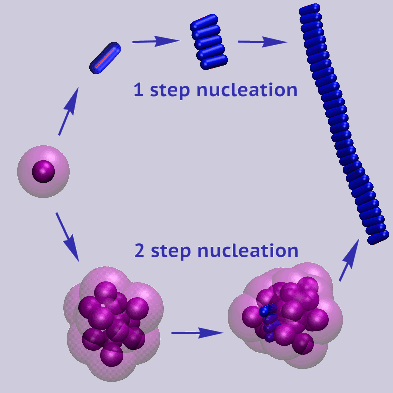
Simulation results show the formation of oligomers and fibrils by a direct nucleation-and-growth
mechanism, by two-step-nucleation through the conversion of an oligomer into a fiber or vice versa,
and by fibril-enhanced cinversion of oligomers into fibrils
[1].
|
|
self-assembly
|
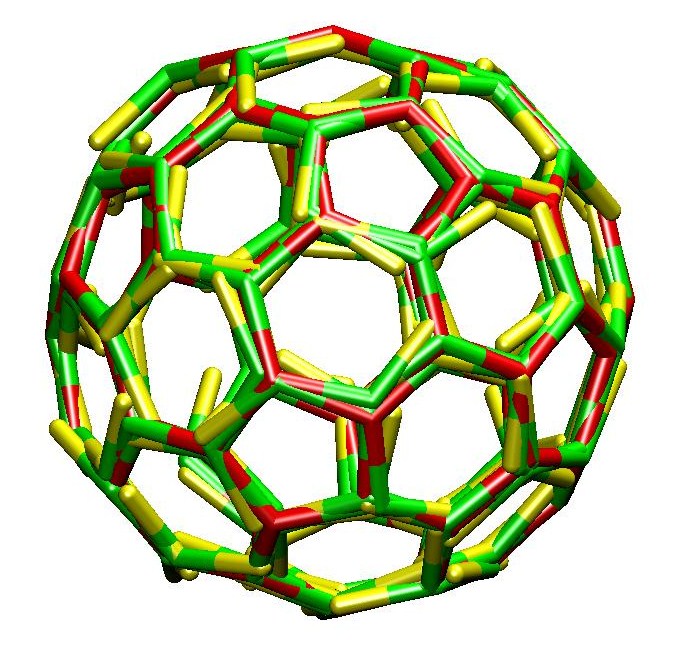
We have presented the first simulations
of the self-assembly of three-legged clathrin proteins
into polyhedral cages.
In living cells
clathrin lattices grow next to the cell membrane,
thereby wrapping the membrane around any external cargo
the cell wants to internalize (endocytosis).
The simulations reveal that the key to cage formation
lies not with clathrin's characteristic shape,
but rather requires leg-leg interactions
with a directionality
[3, 4, 5].
We also made the first estimate of the binding energy
[6].
Click here for a movie (20MB).
|
free energies
|
Intermediate states of reactions are conveniently studied
in molecular dynamics simulations
by suppressing motion along the reaction coordinate.
We introduced the exact relation
between the constraint force
and the free energy profile
[7].
While the remaining internal coordinates are clearly irrelevant,
we were the first to show that they are
not required in the calculation either
[8].
As an example,
these methods
enabled us to determine the free energy profile of
pore formation in lipid membranes
[9,10],
by introducing a novel reaction coordinate
based on the local particle density.
|
.jpg)
|
liquid crystals

|
We have pioneered event-driven Brownian dynamics
[11],
in order to study the flow behaviour of liquid-crystalline
suspensions of elongated hard rods.
The simulations clearly show
the collective tumbling motion of the rods under shear flow,
in good agreement with the motion of the director in experiments
[12].
The simulations reveal that the rods tumble at all shear rates,
and explain why measured directors stops tumbling
at a critical shear rate
[13].
Click here for a movie (29MB).
|
hydrodynamics
|
We have merged molecular dynamics
with stochastic rotation dynamics
[14]
to capture the combined effects of Brownian and hydrodynamic forces
in colloidal suspensions.
This enabled us to study, for the first time,
the sedimentation of hard spheres
[15]
and of attractive colloidal particles
[16],
highlighting the importance of hydrodynamic backflow
and clustering on the mean sedimentation velocity.
These movies show the difference between colloids sedimenting
with (6MB)
and
without (6MB)
hydrodynamics.
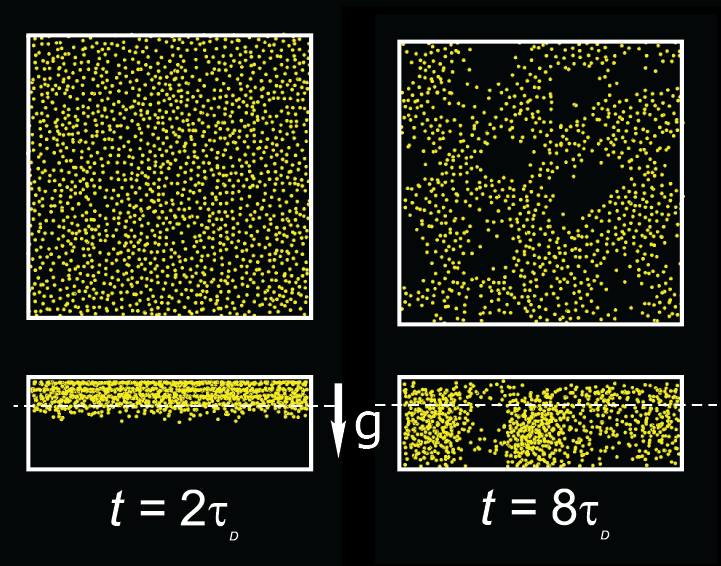
Sedimentation in a planar slit gives rise to pattern formation,
shown on the right in side view (bottom)
and in horizontal cross section (top, at height of dashed line).
|
|
|
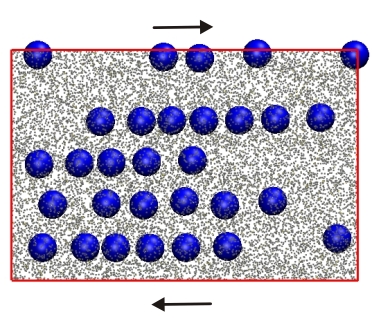
We have developed Responsive Particle Dynamics (RaPiD)
to simulate soft matter at the mesoscopic level,
by modeling complex molecules as single particles
[17].
The crux of the method
is that every particle pair is endowed with an internal coordinate
describing the evolution of the pair interaction,
thereby naturally introducing the transient forces
responsible for the memory effect of viscoelastic fluids
[18].
RaPiD has succesfully been applied to study
shear-banding resins,
shear-fracturing telechelics
[19],
and shear-induced alignment (picture) of colloids dissolved in worm-like micelles,
among others.
Click here for a movie (35MB).
|
polymer dynamics
|
Atomistic molecular dynamics simulations of polymer melts
become exceedingly slow for chains beyond ~100 carbons.
Longer chains require coarse-grained models,
combining ~20 carbons into a single particle,
but the effective interactions between these beads
are so soft that the chains lose their most important feature:
their uncrossibility.
We have developed twentanglement
to preserve uncrossibility in coarse-grained simulations
[20],
thereby recovering excellent scaling laws
(diffusion with power -2, viscosity with power 3.5)
for linear polyethylene upto 1000 carbons
[21].
|
|
|
|
|
 |
|
 |
|
|
| |
|
|
|
|
|
|
|
|
|
selected publications
Protein aggregation
Self-assembly
Free energy
Liquid crystals
Hydrodynamics
Visco-elastic fluids
Polymer dynamics
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
 |
|
|
|